
Pioneering computational methods to calculate diamagnetic susceptibility tensors of organic molecular crystals, have shown that polymorphs of the same molecule can have very different anisotropy in the response to an external magnetic field. The diamagnetic suspceptibility can be estimated by tensorial addition of molecular magnetic tensors, helping account for how strong magnetic fields influence the polymorph that crystallises for some molecules, or conversely, determine when it is unlikely that a magnetic field will not help to find new polymorphs. The unusual mechanism of the reversible phase transitions between desloratadine polymorphs with molecules of different conformations can be understood with phonon calculations, using a methodology being developed to estimate the relative thermodynamic stability of polymorphs as a function of temperature. Unfortunately, the difference in colour that considerably aids polymorph identification in some systems, such as chalcones, is far more sensitive to the quality of electronic structure calculations than the phonons and magnetic properties and cannot be predicted using the band gaps calculated by the popular GGA functionals, routinely used for pharmaceutical crystals. However, these methodological developments of periodic DFT-D calculations are enabling us to collaborate with experimental groups, aiding the prediction, crystallisation and identification of new polymorphs of organic materials and the understanding of crystallisation.
Many organic molecules have more than one crystal form, known as polymorphs. Often, these polymorphs exhibit different physical properties, e.g. solubility, dissolution rates, melting points and mechanical stability. The unexpected crystallisation of a novel polymorph can cause serious problems for the pharmaceutical industry;1 but novel materials with superior properties may also be found through new polymorphs.2 Crystal structure prediction (CSP) is being developed as a valuable computational tool to help the pharmaceutical, agrochemical and other speciality organic materials industries.3 Calculation of the physical properties of computer-generated structures helps in designing crystallisation processes and ensuring quality control in the product. Periodic dispersion-corrected density functional theory (DFT-D), though computationally costly, has shown considerable potential to improve CSP, particularly for flexible pharmaceutical molecules as it treats inter- and intramolecular forces on an equal footing.4
As a research group we have been involved in the development of CSP methods for the last two decades. We have benefited from the Materials Chemistry Consortium which allows us to explore the application of periodic DFT to calculate the properties of polymorphs of conformationally flexible organic molecules containing multiple functional groups. Recently it was demonstrated in coronene that external magnetic field can modify the polymorphic product of a crystallization process.6 Supported by EU’s Horizon 2020 MagnaPharm project, two methods were developed to calculate diamagnetic susceptibility of organic molecular crystals, with one completely based on periodic DFT. We showed that different polymorphs of organic molecules can have drastically different anisotropy in their diamagnetic susceptibility, which points to a possible mechanism where an external magnetic field may affect the crystallization process of an organic molecule.7
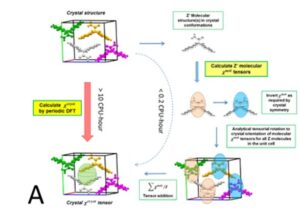
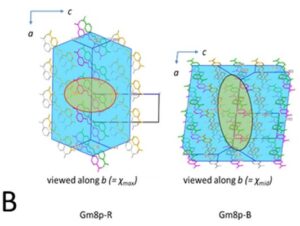
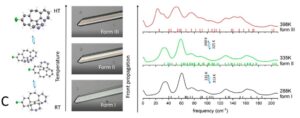
Two methods, periodic DFT-D (red arrow in A) and tensor-addition (blue arrows in A), developed for the calculation of crystal diamagnetic susceptibility, which can differ between polymorphs. E.g. the two polymorphs of Gm8p chaclone, overlaid on their BFDH morphologies of the same molecule, shown in B. C: reversible phase transitions between polymorphs of desloratadine, proceeding as visible wavefronts in single crystals (middle), involves conformation changes of desloratadine molecules (left). On the right hand side, phonon spectra calculated with periodic DFT-D method are overlayed on experimental THz phonon spectra.
Working with CASTEP developers, we developed protocols for the calculation of harmonic phonons in organic molecular crystals, to investigate the relative thermodynamic stabilities of desloratadine, coronene, chalcone and other polymorphs. For the unusual reversible phase changes between the desloratadine polymorphs, which were observed experimentally as wavefronts in single crystals,5 phonon dispersion calculations showed that the phase changes involve conformational changes of the desloratadine molecules, and did not fit the classifications common in inorganic materials. Current applications of periodic DFT-D to organic molecular crystals are sensitive to the quality of the dispersion corrections and extremely expensive for other than GGA functionals. The limitations for calculating band gaps and optical properties was shown by an investigation of the coloured polymorphs of two chalcones.8 The colour differences could be explained by a series of higher quality calculations on the many dimers and stacks of molecules in the crystals. Our collaboration with experimental groups has shown the ability of computational studies to aid the discovery of new polymorphs, and highlighted that further development of computational methods are needed to fully exploit periodic DFT-D in pharmaceutical development. More studies of polymorphic systems are required to understand the factors that control which polymorph is found when the thermodynamic differences are very small. Computationally-aided design of organic molecular products has great potential for aiding the development of new organic materials with carefully controlled physical properties.
Outcomes:
- Developed methods to calculate the diamagnetic susceptibilities of organic crystals;
- Showed that the anisotropy of different polymorphs of the same molecule can be drastically different, pointing to a possible mechanism for external magnetic field to affect the product of crystallization process;
- Established a protocol for calculating the harmonic phonon spectra of pharmaceutical polymorphs, which was used to analyse the mechanism of the reversible phase changes of desloratadine and has been applied to the thermodynamic properties of other polymorphic systems;
- Demonstrated the challenges in accounting for the colour differences between organic polymorphs;
- Convinced leading experimental groups of the value of computational prediction of potential polymorphs and their physical properties to aid investigations into the polymorphic behaviour of pharmaceutical and other organic molecules and to provide a molecular level understanding.
References:
[1] Faraday Discuss. 2018, 211, 441; [2] Nature 2017, 543 (7647), 657; [3] Faraday Discuss. 2018, 211, 9; [4] Acta Crystallographica Section B 2016, 72 (4), 439; [5] Crystal Growth & Design 2020, 20 (3), 1800; [6] Nature Communications 2016, 7; [7] J. Phys. Chem. A 2020, 124 (7), 1409; [8] Crystal Growth & Design 2020, 20 (10), 6346
Collaborators:
UCL Chemistry: Sarah (Sally) L. Price, Rui Guo, Louise Price. Experimental collaborators on MagnaPharm led by Prof Simon Hall, Bristol, and on Templating Project led by Prof Alastair Florence, Strathclyde.
Funding:
European Research Council, Horizon2020 FETOPEN on Magnetic Control of Polymorphism in Pharmaceutical Compounds (MagnaPharm, grant agreement 736899) EUR2.88M 2016 –
EPSRC Responsive Mode EP/K039229/1 2013 on Computationally Designed Templates for Exquisite Control of Polymorphic Form £1,248,345 2013 – 2017
Contact:
Prof. Sarah (Sally) L. Price; s.l.price@ucl.ac.uk