
Formed via aqueous mineral carbonation of Mg2+ ions, the crystallization of magnesite (MgCO3) represents an industrially effective route to CO2 storage and utilisation, generating stable and ready-to-use carbonate-based materials, revenues of which are expected to reach $1 trillion/yr. by 2030 (Accelerating Breakthrough Innovation in CCUS, 2017). Mg sources are widespread and plenty and include Mg-silicate deposits (>100,000 Gt), mine wastes, and alkaline industrial residues. However, CO2 mineralisation is limited by the slow rates of MgCO3 precipitation. Due to the highly hydrated character of Mg2+ in aqueous solutions (ΔhydG° = –456 kcal.mol–1), the difficulty in precipitating magnesite has long been ascribed to the slow kinetics of Mg-dehydration. However, geological records show that anhydrous high-Mg (dolomite, [Ca,Mg]CO3) and sole-Mg carbonates (magnesite, MgCO3) form in weathering of ultramafic rocks (Mg-rich and low silica) at near-surface conditions (T<60 °C), which are very different from the high-T and/or pCO2 necessary to stimulate the synthesis of MgCO3. Natural solutions are far from pure water, being rich in ions, making solution environments highly influential on the rate-determining Mg-dehydration step. Resolving the catalytic role of composition is imperative to determining what catalyses MgCO3 formation in sedimentary environments.
On ARCHER and Thomas HEC, we have conducted a computational investigation based on molecular dynamics (MD) and enhanced sampling metadynamics (MetaD) methods, in addition to selected electronic structure analysis based on density functional theory (DFT) calculations, to provide a theoretical characterisation of the Mg–H2O dissociation reaction mechanism in the presence of additives in solution. We reason that the root of these effects may reside in the ability of organic ligands and inorganic anions that are typically present in aqueous environments to activate Mg-dehydration, and subsequent nucleation and growth steps, by influencing the hydration structure of Mg2+. Our computational characterisation of the Mg–H2O dissociation mechanism shows that the slow kinetics of Mg-dehydration in pure liquid water is due to a highly metastable five-coordinated intermediate, which is inaccessible at low T (Figure A). We show that additives can stabilise undercoordinated Mg2+ hydration configurations, even when these ions are in the second hydration shell of the cation (Figure B). Wavefunction analyses revealed the specific changes in bonding responsible for these equilibrations of five and six coordinate states, with insight into the mechanisms by which these can inter-change and subsequently open-up coordination sites on the central Mg2+ ion towards initiating nucleation.
This work opens the pathway to determine the solution condition promoting CO2 mineralization into added-value Mg-carbonate materials, under mild-T conditions.
On ARCHER and Thomas HEC, we have conducted a computational investigation based on molecular dynamics (MD) and enhanced sampling metadynamics (MetaD) methods, in addition to selected electronic structure analysis based on density functional theory (DFT) calculations, to provide a theoretical characterisation of the Mg–H2O dissociation reaction mechanism in the presence of additives in solution. We reason that the root of these effects may reside in the ability of organic ligands and inorganic anions that are typically present in aqueous environments to activate Mg-dehydration, and subsequent nucleation and growth steps, by influencing the hydration structure of Mg2+. Our computational characterisation of the Mg–H2O dissociation mechanism shows that the slow kinetics of Mg-dehydration in pure liquid water is due to a highly metastable five-coordinated intermediate, which is inaccessible at low T (Figure A). We show that additives can stabilise undercoordinated Mg2+ hydration configurations, even when these ions are in the second hydration shell of the cation (Figure B). Wavefunction analyses revealed the specific changes in bonding responsible for these equilibrations of five and six coordinate states, with insight into the mechanisms by which these can inter-change and subsequently open-up coordination sites on the central Mg2+ ion towards initiating nucleation.
This work opens the pathway to determine the solution condition promoting CO2 mineralization into added-value Mg-carbonate materials, under mild-T conditions.
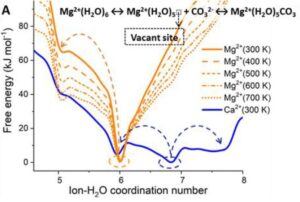
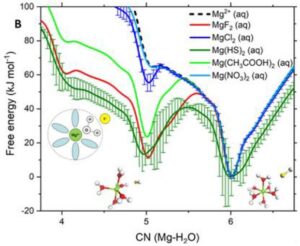
Free energy profiles of hydrated Ca2+ and Mg2+ ions as a function of the ion-water coordination number, obtained from metadynamics simulations at different temperatures. (A) Free energy as a function of the Mg-H2O coordination number, CN(Mg-H2O), for hydrated Mg2+ (no counterions) and solvated Mg2+ with a counterion in the second hydration shell. Five- and six-coordinated states on the free energy profile of Mg(HS)2 (aq).
Outcomes
• Now understand how to design additives to promote Mg-dehydration and the subsequent steps of MgCO3 nucleation and growth;
• Now understand how molecules with different potential energy surfaces can be separated on a surface;
• New links to Cambridge Carbon Capture Ltd;
• Training of two PDRA in the geochemistry using ab initio and classical MD simulations.
Collaborators
Dimitrios Toroz, Fu Song and Greg Chass, Queen Mary University; Encarnacion Ruiz-Agudo, University of Granada; Pedro Alvarez-Lloret, University of Oviedo; Mariette Wolthers, Utrecht University; German Montes-Hernandez, University of Grenoble.
Funding
• Horizon2020 (ACT programme) project No 294766 (EUR 700,810)
• Impact Acceleration Account (£25,000)
Contact
Dr Devis Di Tommaso, d.ditommaso@qmul.ac.uk